
联系我们

电话:025-85860978
手机:18913919581
传真:+86-25-85860962
邮箱:sales@searchbio.com.cn
Msn:searchbio@hotmail.com
行业资讯 首页 > 新闻动态 > 行业资讯
罕见病基因治疗已进入再度繁荣阶段,这是一份最值得收藏的基因治疗行业研究

基因治疗是将正常基因或带有治疗作用的基因通过一定方式导入人体,以纠正或补充基因缺陷及异常所带来的疾病的治疗方案。1972年,美国生物学家T. Friefman及R. Robin在《科学》杂志上发表文章《Gene therapy for human genetic disease?》,提出了基因疗法用于遗传疾病治疗的假设。这一文章被广泛认为具有划时代的前瞻性。此后伴随一系列临床试验的开展,基因治疗的发展经历了“初期探索”、“狂热发展”、“曲折前行”和“再度繁荣”四个阶段。
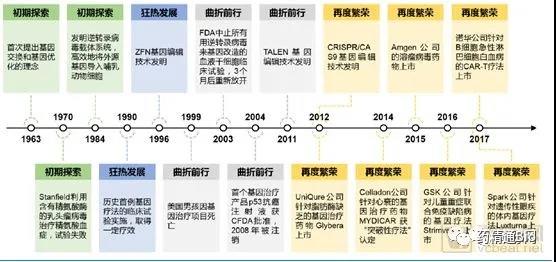
基因治疗发展历史(来源:华金证券)
目前基因治疗有两种基本策略:第一种使用整合型载体,将基因导入前体细胞或干细胞基因组,伴随着细胞的分裂将插入的基因传递至子代细胞;第二种使用非整合型载体,转入的基因独立存在于不具备分裂能力或分裂极慢的宿主细胞核内,以实现相关基因的持续表达。
目前针对这两种治疗策略,基因治疗的具体治疗过程可以分为体外治疗和体内治疗两类。
体外治疗的过程是将患者自体细胞分离,并在体外转入治疗性基因,最后将细胞输注回患者体内。体外疗法一般使用整合型载体,对诸如造血干细胞等具有持续分裂能力的细胞进行基因插入。与之对应的,体内治疗则使用非整合性载体,大多通过静脉注射等与一般药物类似的给药方法,更具可操作性。
在具体应用上,基因治疗技术主要分为以病毒为载体的基因递送技术和基因编辑技术两大类。
基因递送技术可以采用导入正常基因,以缓解自身有缺陷的相关基因,或导入致病基因的抑制性序列如(iRNA、shRNA等)降低致病基因翻译或转录水平等策略。
基因编辑技术则主要以锌指核酸酶(zinc finger nuclease, ZFN)、转录活化因子样受体核酸酶(tranion activator-like effector nuclease,TALEN)和成簇的规律间隔短回文序列重复(clustered regulatory intersoaced short palindromic repeat,CRISPR)-CRISPR相关蛋白(Cas)三类技术为代表,可实现对于目的基因的校正、敲除和增加。
目前对于体外/体内两种治疗策略的安全性考虑核心因素有所不同,体外治疗侧重基因组整合过程中造成插入突变的风险,体内疗法则关注病毒输注所造成的过度免疫反应。
除此之外,质粒DNA、裸DNA等也可通过非病毒基因递送方式,诸如显微注射、基因枪或者脂质体、纳米颗粒等。但这种方式可使用的组织/细胞较为局限,目前转染效率较低,相关载体依然在不断开发中。
遗传病、罕见病治疗是基因治疗的重要应用方向,目前已经获批上市的基因疗法多围绕这两点展开。但价格高昂,监管有待完善,竞争激烈但适应症有限,基因治疗行业在业界广泛关注的同时也伴随争议。
接下来,本文将从罕见病、遗传病基因治疗的相关进展,基因治疗相关政策、行业规范及支付体系,竞争格局等三个方面,带你了解不一般的基因治疗。
罕见病、遗传病基因治疗的相关进展
基因疗法目前的应用包括单基因遗传病、血友病、眼科及部分神经退行性疾病,发病/治疗机制相对明确的多基因遗传病以及癌症的治疗研究。本文就第一部分治疗领域的药物获批及临床试验进展情况进行了一定的整理和分析。
已经获批的基因治疗产品
自2016年以来,欧盟EMA和美国FDA共批准了6款基因疗法产品,其中包括两款治疗B细胞血液肿瘤的CAR-T细胞疗法以及四款治疗严重单基因遗传病的基因疗法。
第一款获得批准的AAV基因疗法为荷兰uniQure公司的Glybera,于2012年获得EMA批准,用于治疗脂蛋白脂肪酶缺乏症。但由于其效果有限、定价太高(平均一次疗法100万美元)且适应症太过罕见(发病率1/100万),上市至今仅有一位患者接受治疗,Glybera在2017年黯然退市。
2016年5月,EMA批准了GSK公司的Strimvelis上市,用于治疗腺苷酸脱氢酶(ADA)缺失导致的严重联合免疫缺陷(SCID)(GSK于2018年4月将包括Strimvelis在内的罕见病基因疗法管线以19.9%的股权交换转移至Orchard Therapeutics公司)。
由于免疫系统严重受损,部分ADA-SCID患者会在婴幼儿时期死亡,其他的患者也只能终生生活在无菌环境中,ADA-SCID也因此被称为“泡泡男孩病”。在该疗法出现之前,患者只能依靠干细胞移植或酶替代疗法控制,酶替代疗法需要每周注射,每年价格40万美元左右。
Strimvelis是一种体外基因疗法,使用γ-逆转录病毒将正常ADA基因导入患者CD34+ HSPCs中。该疗法的价格大约在66万美元一次,理论上仅需一次治疗即可终生获益。
2017年12月,FDA批准了Spark Therapeutics公司开发的Luxturna,用于治疗RPE65双等位基因突变导致的遗传性视网膜疾病先天性黑朦症Ⅱ型(Leber’s congenital amaurosis, LCA)。RPE65基因突变导致其蛋白失去异构酶活性,引起了11-顺-视黄醛的缺乏,从而造成患者体内的光感受器细胞无法感光。到了晚期,11-顺-视黄醛的缺乏可能引发视锥细胞和视杆细胞变性,发生严重的视力丧失。
在Luxturna获批上市前,LCA尚无有效治疗药物。Luxturna使用AAV2携带RPE65基因,采用在视网膜下腔直接注射的方法将病毒导入眼球中。该疗法的双眼治疗价格在85万美元左右。
2019年5月,FDA批准了诺华公司旗下AveXis Inc.的体内基因治疗药物Zolgensma,用于治疗SMN1双等位基因缺陷导致的脊髓型肌肉萎缩(SMA)婴儿患者(<2岁)。SMA的发病率约为1/5000-1/12000,表现为进行性肌肉无力、瘫痪等症状。
按照SMN基因拷贝数以及蛋白表达水平,SMA严重程度可分为四种亚型,其中I型最为严重,出现症状时,婴儿一般6个月大,仅有8%的婴儿能够生存超过两岁。在Zolgensma获批之前,美国药企Biogen的Spinraza是唯一被批准治疗SMA的疗法。与Zolgensma的治疗思路不同,Spinraza是一种SMN基因的反义寡核苷酸(ASO),需要通过腰穿鞘内注射,每4个月一次。
Spinraza理论上可能需要终生注射,相对应的治疗价格为第一年75万美元,此后每年37.5万美元。Zolgensma通过静脉输注载有SMN功能基因的AAV9载体,实现对于SMA的根治。该产品官方定价为212.5万美金一次治疗,被称为史上单次治疗最贵药物。
然而在FDA批准上市后的一个月,Zolgensma的研发生产公司AveXis主动向FDA和其他监管机构承认,这一基因疗法在一项动物实验中存在部分数据操纵的问题。
2019年8月6日,FDA启动对于Zolgensma数据准确度的全面审查,截至目前,调查结果未显示该产品在安全性、功效性和质量上存在问题。
2019年6月,EMA对蓝鸟生物开发的药物Zynteglo进行了有条件批准,同意Zynteglo用于治疗12岁以上非β0/β0基因型输血依赖型β-地中海贫血(TDT)患者。TDT是一种遗传性贫血病,由于编码β-珠蛋白的基因发生缺陷或突变,导致β-珠蛋白合成降低而α-珠蛋白过量并以不可溶的包涵体存在于红细胞中,诱导红细胞凋亡产生贫血症状。该病世界范围内发病率为1/10万,欧盟区为万分之一,而部分地区发病率极高(塞浦路斯 14%、撒丁岛10.3%)。TDT的常规疗法为长期输血联合贴螯合剂或造血干细胞移植。
Zynteglo为一种体外基因疗法,通过慢病毒载体将功能性人βA-T87Q-珠蛋白基因导入CD34+ HSC中并回输,使患者能自主生成β-珠蛋白,从而产生足够的血红蛋白,减少或消除输血治疗的必要性。此外,蓝鸟生物正在测试Zynteglo在镰刀型红细胞贫血中的疗效,计划2020年在美国进行销售。该疗法的180万美元一次治疗,是仅次于Zolgensma的“第二贵”药物。
基因治疗临床试验情况分析
截至2019年9月,以lentivirus为载体的体外基因治疗临床试验共有115项,以rAAV为载体的体内基因治疗临床试验共有190项。以ZFN、TALEN和CRISPR-Cas技术作为基因编辑工具的临床试验共32项,大部分针对肿瘤的治疗。
在罕见病/遗传病治疗领域,有4项研究是使用rAAV作为载体的体内基因编辑试验,另外5项为体外基因编辑试验(未使用慢病毒作为基因编辑工具的递送载体)。在正在进行中的基因治疗临床试验中,约58%为Ⅱ期研究、33%为Ⅰ期研究,Ⅲ期临床试验占比大约9%。
目前正在进行的以慢病毒为载体的体外基因疗法临床试验共有55项,多数集中于X-连锁严重联合免疫缺陷、镰刀型红细胞贫血、血友病、粘多糖贮积症(MPS)以及溶酶体贮积症等遗传病的治疗。部分相关临床试验的情况整理如下:
罕见病体外基因疗法部分临床试验整理

数据来源:Clinicaltrials.gov, 探针资本整理
1、rAAV基因递送治疗临床试验概况
在不同血清型中,研究最为充分的AAV2和AAV8最多地被用于临床试验。由于rAAV的不同血清型天然存在对于特定组织和器官的靶向性,目前大多数基于rAAV的基因治疗集中于对肝脏、横纹肌和中枢神经系统的特异性靶向。
多种天然AAV衣壳蛋白均可传输至肝脏,该载体在治疗A型血友病、B型血友病、家族性高胆固醇血症、鸟苷酸转糖化酶缺乏症和Crigler-Najjar综合症中发挥作用。
AAV8和AAV9可以针对全身多种肌肉类型,用于治疗杜氏肌营养不良(DMD),以及运用心脏中与某些信号通路和代谢相关的特定基因如SERCA2a治疗心脏衰竭等。

AAV为载体的体内基因疗法临床试验统计(截至2018年11月)(来源:Nature)
从上图可以看出,临床开发中rAAV的基因递送治疗中,很大一部分作用于中枢神经系统,包括脑以及眼球。眼球是一个相对分隔的器官,血眼屏障的存在使得rAAV病毒直接眼底注射即可达到基因递送的效果,且治疗所需病毒载体数量较少,不会导致炎症性免疫反应,减少了由于抗体中和或免疫反应引起的疗效降低。
目前针对眼部进行基因治疗临床试验的适应症包括先天性黑朦、遗传性脉络膜视网膜营养不良、全色盲和Leber遗传性视神经病变(LHON)。
相比之下,大脑更加复杂,体积相对较大,因此脑实质直接注射rAAV会导致rAAV的局部分布。
对于如帕金森症这类致病区域相对明确的神经系统疾病,核壳区域使用rAAV即为理想治疗方法。另外,鞘内注射病毒至脑脊液间隙也可获得更广泛的中枢神经分布,但相对风险较大。
AAV9和AAVrh.10可以穿越血脑屏障,因此可用其将基因传递至神经元和胶质细胞。目前已经有研究证明了全身性rAAV治疗中枢神经系统疾病的疗效,包括脊髓型肌萎缩症(SMA)、肌萎缩侧索硬化症、卡纳文病等适应症。部分rAAV基因递送临床试验的情况如下表所示:
部分rAAV基因递送临床试验统计
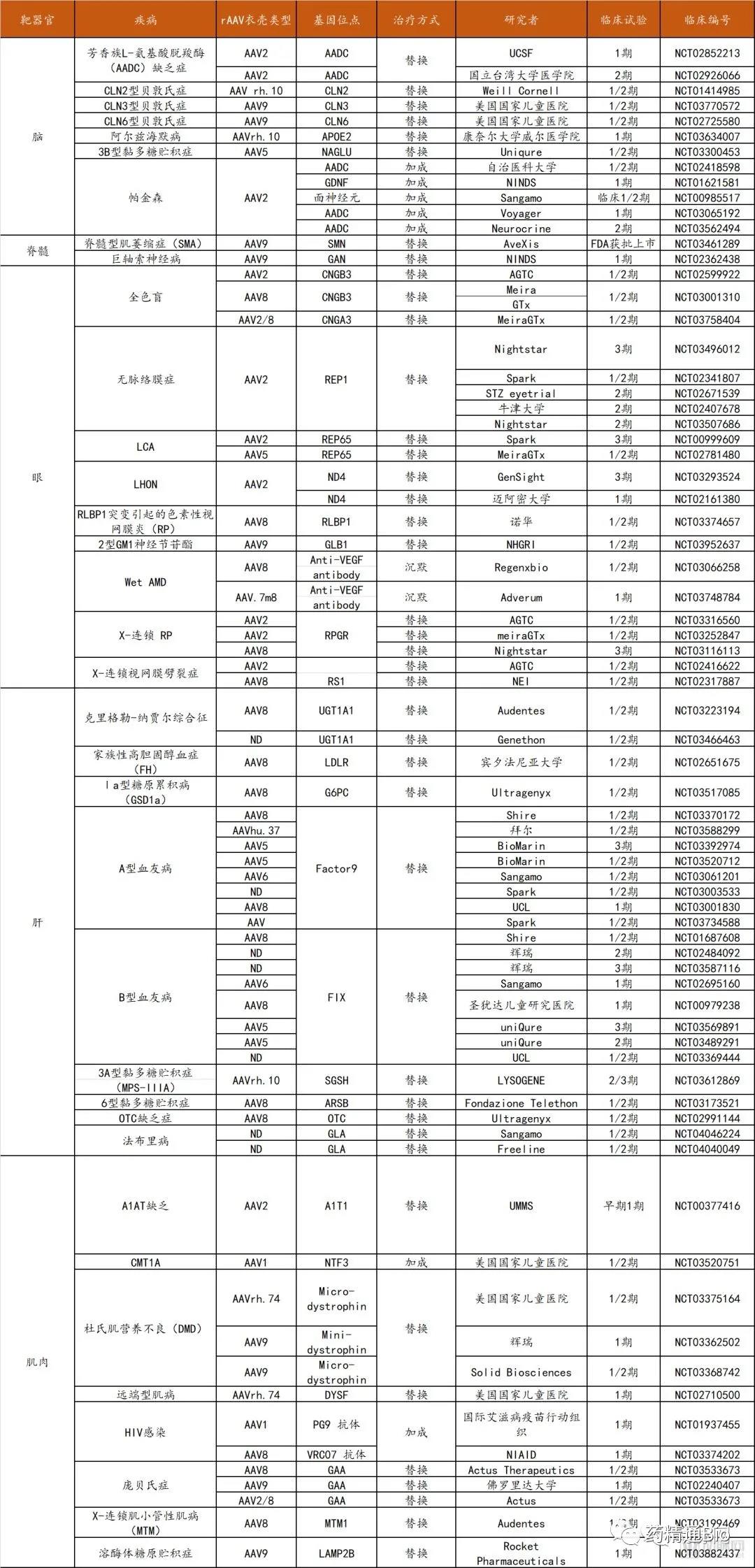
数据来源:Clinicaltrials.gov,探针资本
2、基因编辑临床试验汇总
由于基因编辑技术本身较新,机制并非完全揭示,脱靶效应并非完全可控,出于安全性的考虑,即便在科研领域引起了极大的关注和广泛的研究,基因编辑技术真正在临床当中应用仍然相对有限。
截至目前,在临床试验中可查的三种基因编辑技术分别为ZFN (14项),TALEN( 3项),CRISPR (15项),其中ZFN的大部分临床试验针对HIV感染(8项试验,CCR5作为靶点),TALEN和CRISPR的大部分临床试验集中在多种癌症的治疗领域(13项),并且中国的临床试验总数在全球名列前茅。
在罕见病/遗传病治疗领域方面,ZFN的5项试验全部被Sangamo公司囊括,适应症包括B型血友病、MPS以及地中海贫血和镰状细胞病(与Bioverativ赛诺菲子公司合作进行)。
CRISPR-Cas9相关试验共四项,3项为针对血液疾病的体外治疗,其唯一的体内治疗研究针对先天性黑朦。
罕见病/遗传病基因编辑临床试验汇总
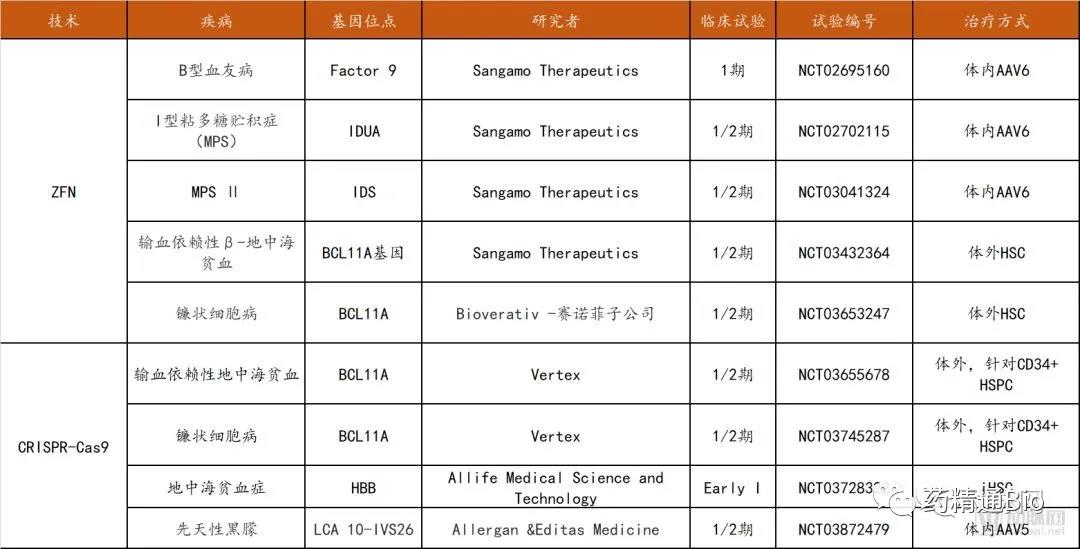
数据来源:clinicaltrials.gov,探针资本

首例CRISPR体内基因编辑药物治疗原理(来源:Editas官网)
基因治疗相关政策、行业规范及支付体系
基因治疗,尤其是采用基因编辑技术进行的基因治疗作为一项新兴技术,在世界各个地区受到不同政府组织不同程度的监管。其中欧美地区在该领域具有相对严谨和完善的监管体系,国内法律法规及监管则有待进一步的发展。此外,行业内多个国际联合组织同样在规范技术及其临床应用上发挥着重要的作用。
具体到罕见病的基因治疗方面,目前70%的基因疗法都是针对罕见病。美国、欧盟、日本、以及中国等多个国家和地区在近些年均出台了一系列针对罕见病的利好政策,如税收抵免、专项基金资助研究、市场独占期、加快其注册审评审批等。
罕见病疗法在申报临床试验时还可适当的减少临床试验病例数或者提出免做临床试验申请。比如Mustang Bio公司和圣犹达儿童研究医院合作开发的针对X-SCID的MB-107慢病毒载体基因疗法就被美国授予再生医学先进疗法认定(RMAT)。
RMAT是专门为再生性疗法设立的审评途径,与FDA的突破性疗法认定类似.这一认证将加快该疗法的开发和评审速度。
2017年12月先天性黑朦治疗药物Luxturna在美国上市后,我国药监局在2018年11月1日将其列入第一批临床急需境外新药公示名单,目前正在通过快速审批通道。
由此可以看出,针对罕见病的政策在大幅度倾斜,这也鼓励了基因疗法针对罕见病治疗领域的引进、研发、生产。此外,由于目前上市遗传病基因治疗产品超高的定价,该疗法的支付体系亦是多方关注的热点。
美国基因治疗政策梳理
美国是基因治疗方面先驱者,对基因治疗的监管体系相对完整。与世界各国的相关监管机构相比,美国倾向于保守和谨慎的态度。比如欧盟于2012年批准了第一例基因治疗药物,而FDA直到2017年才批准第一款基因治疗药物。
早在1974年美国国立卫生研究院(National Institutes of Health,NIH)就成立了重组DNA咨询委员会(Recombinant DNA Advisory Committee,RAC),其使命从对于使用操纵核酸的新兴技术开展的研究,扩大到了涵盖对人类基因治疗方案的审查和讨论。1984年,FDA首次对基因治疗产品进行监管,形成了FDA、NIH对基因疗法双监管的格局。1990年,FDA开始批准人类基因疗法试验。但1999年基因疗法出现了死亡事故,这导致政府加强了对基因治疗的审查监管力度,FDA和NIH先后公布了基因治疗的多项监管方法,同时具体监管方式也在不断发生改变。
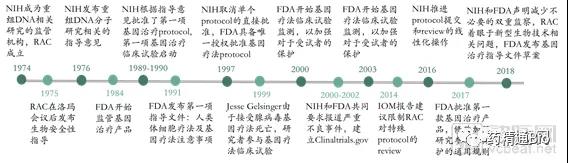
美国对基因治疗的监管历史(来源:NEJM,探针资本)
2018年8月,FDA和NIH共同在《NEJM》上发表文章,声明并没有充分的证据证明基因疗法存在特殊的、不可预知的安全风险,不需要不同于其他治疗方法的监管措施。
因此,美国将对现有基因治疗的监管体系实施改革,监管职能逐步精简到单一机构FDA,鼓励基因疗法的开发。FDA根据基因治疗技术发展情况的不断更新,从产品研发的各个环节给予企业指导,目前共发布了12个指南,涉及七个方面。
FDA关于基因治疗的相关法规
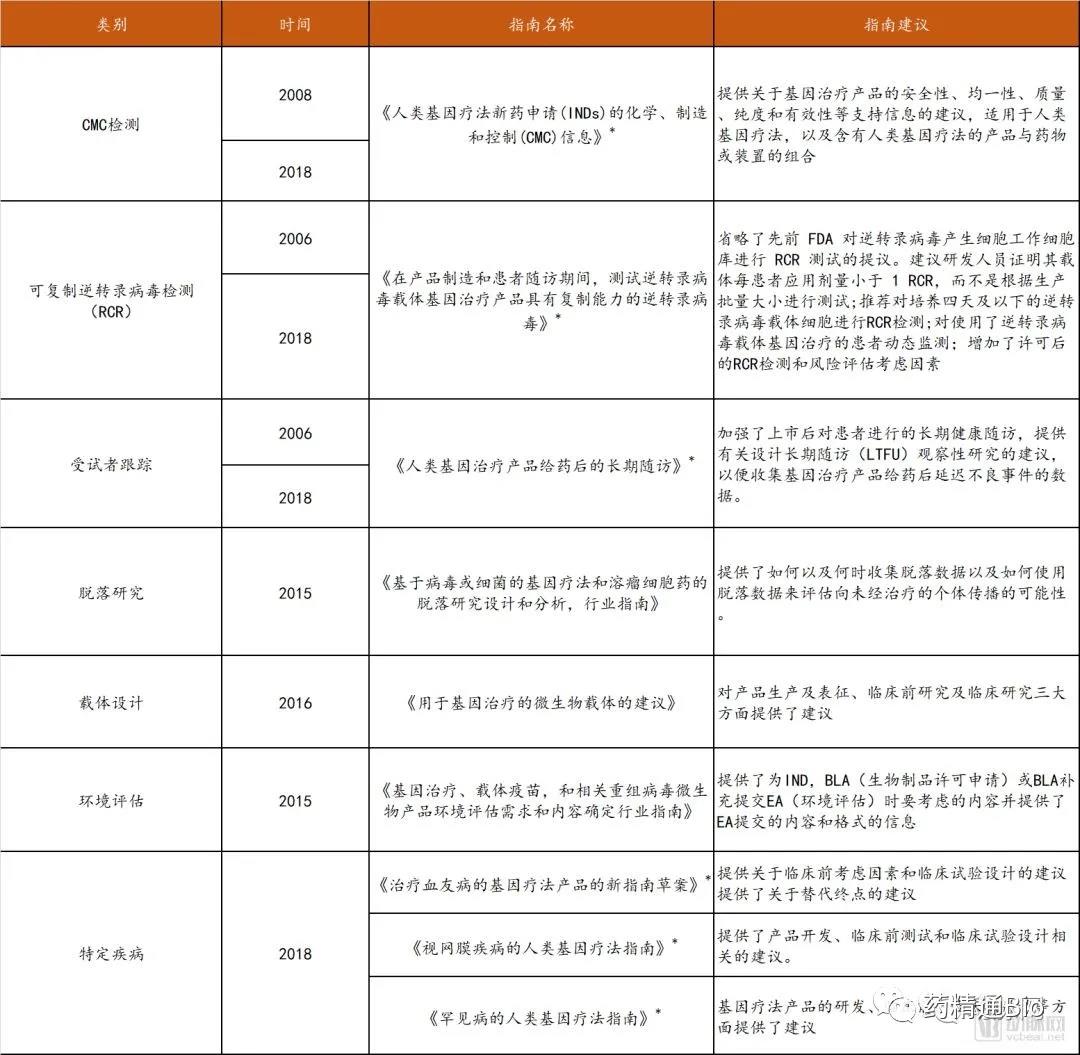
* 代表草案
数据来源:FDA官网,探针资本整理
以2018年发布的《罕见病的人类基因疗法指南》(草案)为例,该指南旨在帮助申办者设计临床开发计划,就其可能存在有限的研究人群、规模,潜在的可行性和安全性问题,以及与解释有效性的问题给出了建议。
国内基因治疗政策梳理
在基因治疗领域,我国在基础研究和临床试验开展相对较早,1991年便进行了2例逆转录病毒体外治疗B型血友病的临床试验。然而与之对应的是长久以来政策法规相对落后、内容也相对简单,对其研究过程中涉及到的具体问题未有详细的说明和规定,法规约束性不强。
早期,国内在此类产品的审批上相对松懈,比如2003年一款名为“今又生”的针对头颈部肿瘤P53基因抗癌药物,在临床试验仅有100余例样本且没有标准Ⅲ期临床试验数据的情况下,就被获批上市,宣称“全球首例获批上市的基因治疗药物”;此外还有2018年底让全球为之震惊的“世界首例基因编辑婴儿”事件,也严重损害了中国科技界及相关政府机构的形象和利益。
在这样的背景下,2019年起,相关政府机构开始不断加强对于基因治疗领域的监管:
2019年4月发布的《民法典人格权编(草案二次审议稿)》增加规定:从事与人体基因、人体胚胎等有关的医学和科研活动的,应当遵守法律、行政法规和国家有关规定,不得危害人体健康,不得违背伦理道德。在8月22日提交的人格权编草案三审稿中进一步增加规定:从事此类活动“不得损害公共利益”。2019年5月发布《中华人民共和国人类资源遗传管理条例》,在基因编辑方面根据最新形势变化做出了相应的修改和完善。
国家药典委2017年立项,参考FDA、美国药典等对于基因治疗制品的要求,于2019年6月公布了《人用基因治疗制品总论(公示稿)》,对通用的技术提出了要求,为保障制品的安全性、有效性奠定良好基础,也将推动此类技术制品的产业化进程和临床应用。此外,国务院表示,为了进一步加强对包括“基因编辑”在内的生命科学研究、医疗活动的规范和监管,2019年还将加快生物技术研究开发安全管理和生物医学新技术临床应用管理方面的立法工作,以形成相关领域的全过程监管链条。相信基因疗法的相关技术在未来将会以更严谨的方式在国内发展。目前国内涉及基因疗法的相关部分法律法规整理如下。
国内基因治疗相关部分法规

数据来源:公开资料,探针资本整理
基因治疗相关行业规范
除了相关政府部门的监管和指导之外,针对基因治疗这一新型疗法,国际上同样存在相关的行业联合会,比如NIST基因编辑协会、国际标准化组织ISO以及再生医学联盟(ARM)等。联合会在科学家和相关企业的呼吁下,为基因治疗制定生物伦理国际框架。
ARM是细胞和基因治疗以及更广泛的再生医学领域国际倡导组织,近期主导开发了基因治疗开发人员原则声明。
《原则声明》明确了使用基因编辑技术的五项关键原则,包括认可体细胞基因编辑的治疗应用研究,支持使用基因编辑标准以促进安全有效的基因编辑疗法;呼吁继续发展个国家和区域调控框架,以指导体细胞基因编辑技术的发展;暂停生殖系基因编辑目前在人体临床环境中的使用以及除非有关生殖系基因编辑的伦理和潜在安全问题得到充分解决,否则不支持或宽恕生殖系基因编辑用于人类临床试验或人类植入的共同承诺。
该声明由13个使用基因编辑技术的共同签署,包括已有基因治疗产品上市的蓝鸟生物,华人科学家张锋创立的Editas Medicine以及ZFN技术垄断巨头Sangamo Therapeutics等知名企业。
支付体系
几款针对遗传病开发的基因治疗药物陆续获批上市,其单次治疗所需的费用不断刷新着人们的认知。史上最贵药物榜单的前三名都被基因治疗药物所占据:最贵药物为诺华旗下Zolgensma,单次费用212.5万美元;第二是蓝鸟生物Zynteglo,单次180万美元;第三是Spark Therapeutics旗下Luxturna,单次85万美元双眼。
虽然随着相关药物的进一步获批,在打破寡头的市场独占性后会迫使寡头降价,但基因治疗药物本身在研发、制造工艺以及用药后对患者进行持续监测上的成本都决定了该疗法的价格在短期内仍保持在高位。因此如何支付这类价格高昂的药物,仍是一个难题。
现阶段采取的方式包括支付方和药企共同协商,药企提出了分期付款、延长付款、按疗效付费等创新付费方式。
比如蓝鸟生物表示Zynteglo的全款可以在五年分期,每年支付约357567美元,患者在开始治疗的时候先支付20%的一次性基因治疗费用,另外80%将取决于治疗成功率。诺华同时在与保险公司达成“基于结果的协议”,商讨五年时间内的支付模式。
诺华和蓝鸟等基因疗法药企提供了基于价值的定向选择,即药物在起效或分阶段几年后才接受全价。但目前美国的医疗补助和医疗保险制度并非为这些新方案而设立,患者在接受基因治疗后的支付问题上,依然会遇到多种挑战。
因此,罕见病基因疗法的支付改革急需进行。我们对目前上市的部分罕见病基因治疗药物的支付方式、政策以及限制进行了梳理,详见下表。
罕见病基因药物支付情况梳理
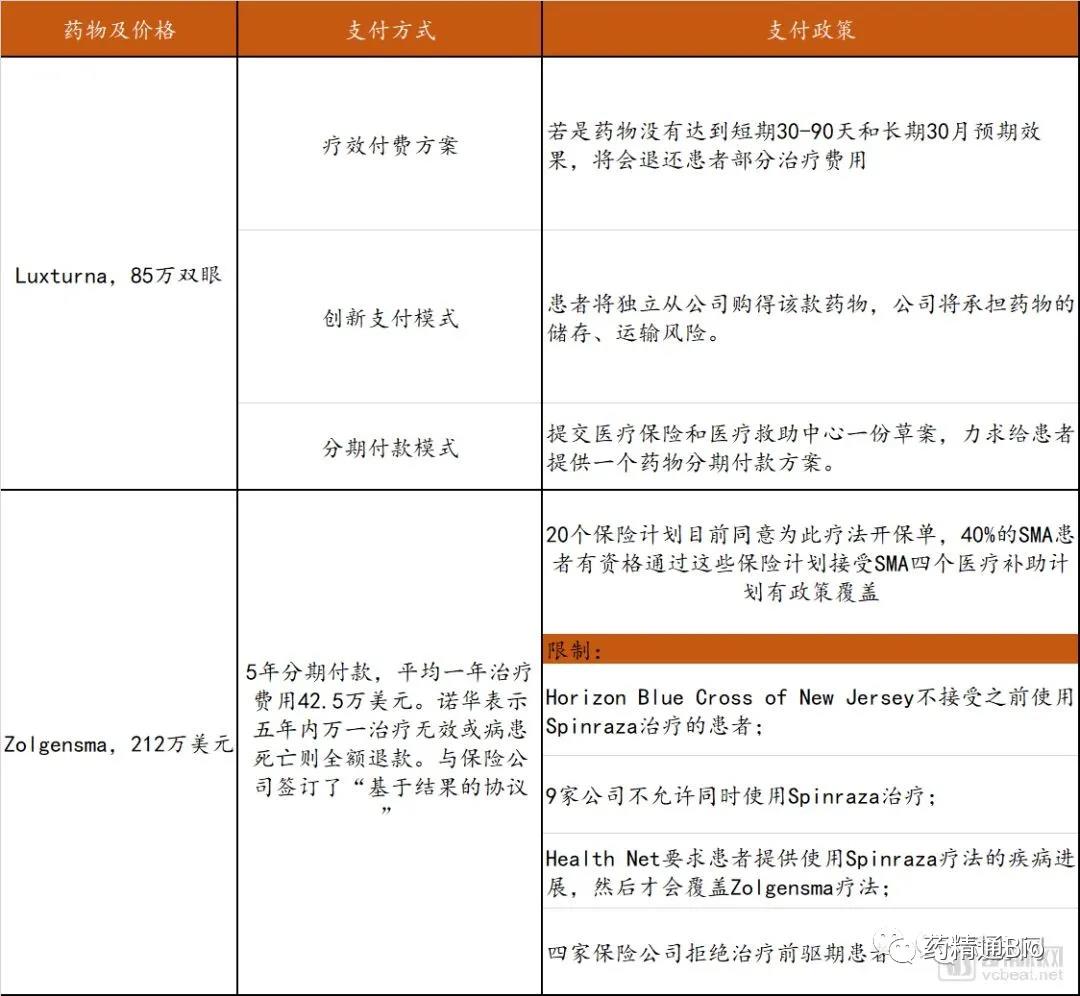
数据来源:FiercePharma、探针资本整理
参考各国罕见病保障体系,美国采取以商业保险为主、政府医疗保健计划为补充的形式进行补助。患者可以通过商业保险公司或者药品保险经营者自愿参加处方药计划,根据药品目录下不同的层次、不同层级的药品共付数额及报销情况灵活选择。
其他国家罕见病的医疗保障体系如下图所示。考虑到罕见病基因疗法能够达到的“一次治疗、终生治愈”的潜在疗效,在相关疗法价格进一步下降的前提下,相比于传统疗法,基因治疗给社会带来的总的经济负担有可能反而减小。
因此,这里并不排除国家政府或医疗补助项目在这一领域给予资金支持的可能性。
综上,我们推测,罕见病基因疗法的支付方式,或许会从现有的按时间收费、按疗效收费、保险公司付费发展为多支付主体、多支付手段相结合的方式。
我国的罕见病的保障体系与上述国家相比较为单一,主要是通过医保报销。国内涵盖罕见病的商业保险产品较少,相关保险保费较高、赔付条件较为苛刻。我国一些省市地区(如上海、青岛、浙江)正在探索并出台具有地方特色的罕见病药物保障模式。
总的来看,加强医保谈判争取罕见病基因治疗药物进入医保目录,并建立多方参与、多方筹资的支付体系是我国未来罕见病基因疗法保障体系的发展方向。